
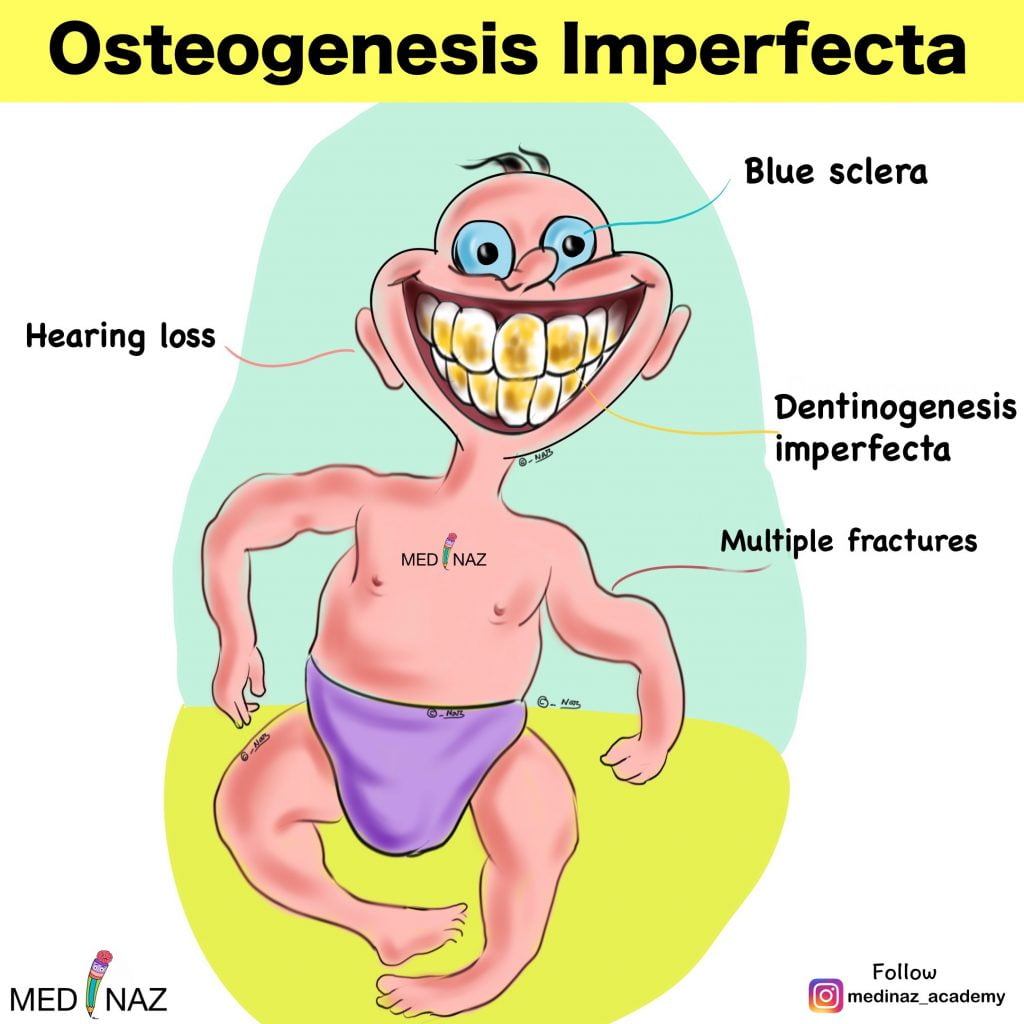
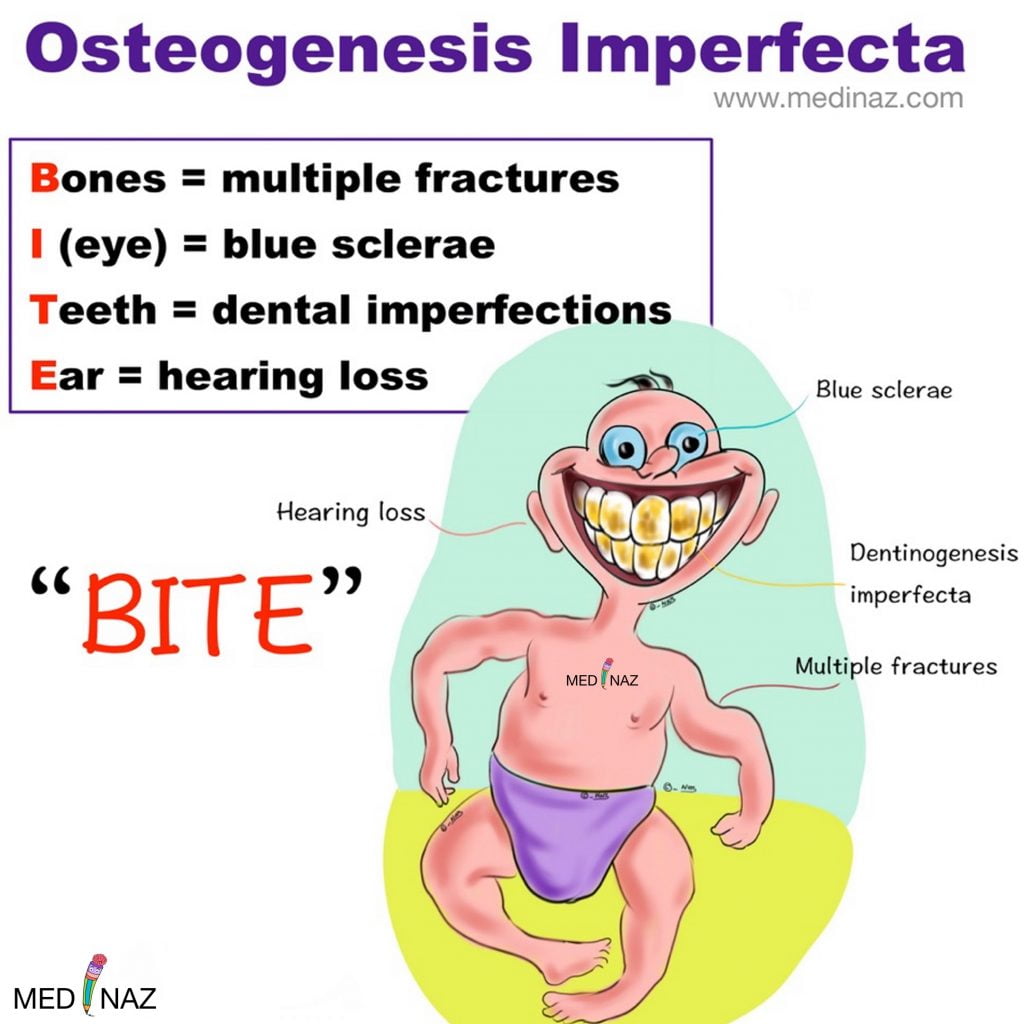
Osteogenesis imperfecta also called “Brittle Bone Disease” comprises a heterogeneous group of heritable disorders. The disorders are frequently associated with blue sclerae, dental abnormalities (dentinogenesis imperfecta), progressive hearing loss, and a positive family history.
Most patients have mutations in one of the two genes coding for Type I collagen
More than 90% of cases exhibit an autosomal dominant inheritance
Commonly mutated genes are COL1A1 and COL1A2
This condition represents one of the most common heritable bone diseases, with a worldwide prevalence of approximately 6 to 7 per 100,000 population.
Classification of Osteogenesis Imperfecta
| Phenotype | Type | Typical features | Inheritance | Gene/Protein defect | Protein defect |
| Non-deforming form | OI type 1 | Mild to moderate bone fragility, normal or near normal stature, in most, blue sclerae, normal dentition in most hearing loss in ~50%. | AD | COL1A1 COL1A2 | Collagen I haploinsufficiency |
| Perinatally lethal form | OI type 2 | Extreme bone fragility, short stature, long bone bowing | AD AR | COL1A1 COL1A2 CRTAP LEPRE1/P3H1 PPIB/CYPB | Collagen I structural mutations Collagen post-translational modification and folding machinery |
| Progressively deforming OI | OI type 3 | Moderate to severe bone deformity, blue sclerae at birth, hearing loss and abnormal dentition common. | AD AR | COL1A1 COL1A2 CRTAP LEPRE1/P3H1 PPIB/CYPB FKBP10/FKBP65 PLOD2/ LH2 SERPINH1/HSP47 CREB3L1/OASIS SEC24D BMP1 WNT1 SERPINF1/PEDF TMEM38B/TRIC-B SP7/OSX | Collagen I structural mutations Collagen post-translational modification and folding machinery Protein folding/endoplasmic reticulum stress sensor COPII vesicle component collagen secretion Proteolytic removal of procollagen N-propeptide Wnt cell signaling pathway Signaling and collagen binding protein, important for mineralization Endoplasmic reticulum cation channel. Calcium homoestasis/collagen synthesis Transcription factor, bone formation |
| Common Variable OI with normal sclerae | OI type 4 | Mild to moderate, bone fragility, normal sclerae, variable dentition, hearing loss in <10%. | AD AR | COL1A1 COL1A2 WNT1 CRTAP FKBP10/FKBP65 PPIB/CYPB SERPINF1/PEDF SPARC SP7/OSX | Collagen I structural mutations Wnt cell signaling pathway Collagen post-translational modification and folding machinery Signaling and collagen binding protein, important for mineralization Collagen binding/extracellular matrix assembly Transcription factor, bone formation |
| OI with calcification of the interosseous membranes | OI type 5 | Calcification of the interosseous membranes in forearm and legs and/or hypertrophic callus. Variable bone deformity, normal sclerae and dentition. | AD | IFITM5 | Transcription factor, bone formation |
| Bruck syndrome type 1 | BRKS1 | Contractures with pterygia, fractures in infancy or early childhood, postnatal short stature, severe limb deformity, and progressive scoliosis | AR | FKBP10/FKBP65 | Collagen folding machinery |
| Bruck syndrome type 2 | BRKS2 | As for Bruck syndrome type 1 | AR | PLOD2/LH2 | Collagen post-translational modification of lysine |
Incidence
Osteogenesis imperfecta TYPE I – 1 in 15,000–20,000 births
Osteogenesis imperfecta TYPE II – 1 in 60,000
Clinical features
Skeletal Defect:
Type I OI –
- The fragility of bones may be severe enough to limit physical activity or be so mild that individuals are unaware of any disability
- Radiographs of the skull in patients with mild disease may show a mottled appearance because of small islands of irregular ossification
Type II OI –
- Continuously beaded or broken ribs and crumpled long bones (accordina femora) may be present
- The long bones may be either thick or thin
Type III & IV OI –
- Multiple fractures from minor physical stress can produce severe deformities
- Kyphoscoliosis can impair respiration, cause cor pulmonale, and predispose to pulmonary infections.
- “Popcorn-like” deposits of mineral in x-rays of the ends of long bones
- Progressive neurologic symptoms may result from basilar compression and communicating hydrocephalus
Type V OI –
- Presence of dislocated radial heads and hyperplastic callus formation
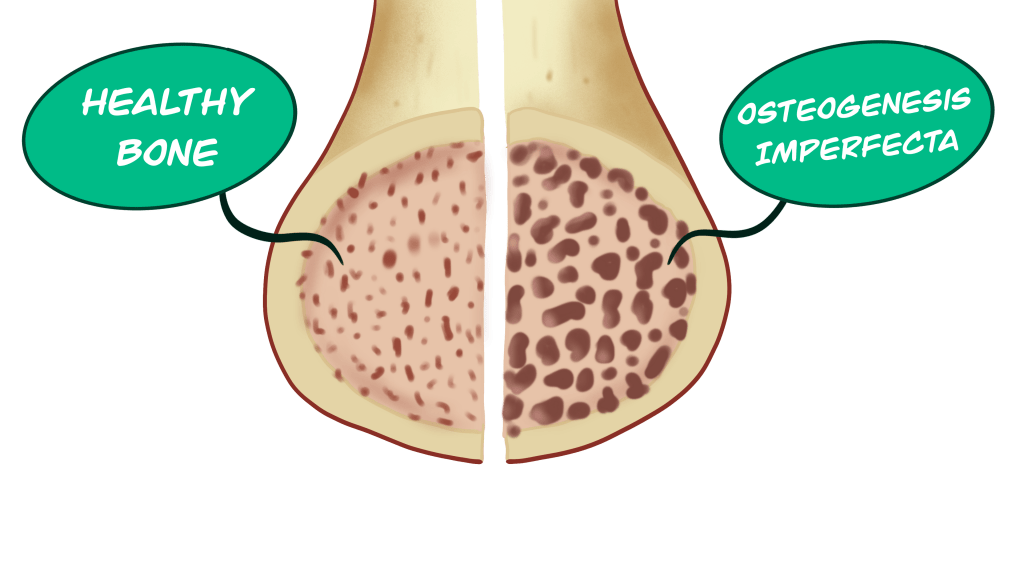
In all forms of OI, bone mineral density is decreased
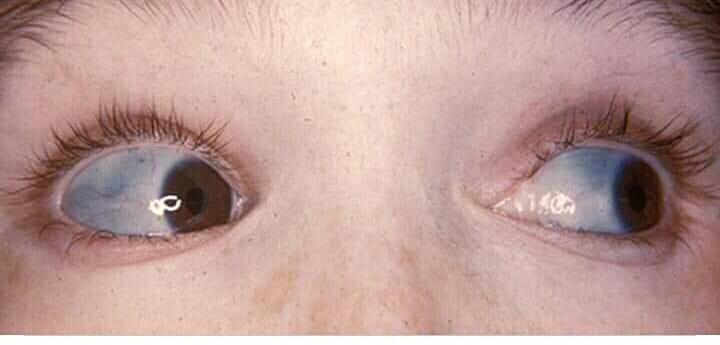
Ocular Features:
- The sclerae can be normal, gray, slightly bluish, or bright blue.
- Blue sclerae, however, are an inherited trait in some families who do not have increased bone fragility
Dental abnormalities:
- The teeth may be normal, moderately discolored, or grossly abnormal. The enamel generally appears normal, but the teeth may have a characteristic amber, yellowish brown, or translucent bluish gray color because of a deficiency of dentin that is rich in Type I collagen.
- The deciduous teeth are usually smaller than normal, whereas permanent teeth are frequently bell-shaped and restricted at the base.
- Radiographs typically reveal premature pulpal obliteration
- In some patients, the teeth readily fracture and need to be extracted. Similar tooth defects, however, can be inherited without any evidence of OI.
- Despite exhibiting similar tooth alterations, osteogenesis imperfecta and dentinogenesis imperfecta are distinct diseases resulting from different mutations. Therefore, the dental defects associated with the systemic disorder osteogenesis imperfecta should be designated opalescent teeth, whereas the term dentinogenesis imperfecta should be reserved for patients with alterations isolated to the teeth
Hearing Loss:
- Hearing loss usually begins during the second decade of life and occurs in >50% of individuals aged >30.
- The loss can be conductive, sensorineural, or mixed, and it varies in severity.
- The middle ear usually exhibits maldevelopment, deficient ossification, persistence of cartilage in areas that are normally ossified, and abnormal calcium deposits
Other Features:
- Joint laxity with permanent dislocations
- Occasional cardiovascular manifestations such as aortic regurgitation, floppy mitral valves, mitral incompetence, and fragility of large blood vessels.
- Some patients develop bouts of a hypermetabolic state with elevated serum thyroxine levels, hyperthermia, and excessive sweating
Diagnosis
Diagnosis requires correlation of the clinical features, radiographic and/or prenatal ultrasound findings, and family history.
For diagnostic confirmation, genetic testing is more sensitive than electrophoresis for type I collagen secreted by cultured dermal fibroblasts.
Bone biopsy may be helpful in select cases.
Serum concentrations of vitamin D, calcium, phosphorus, and alkaline phosphatase usually are normal, although occasionally the latter may be slightly elevated
Treatment and Management
Treatment is aimed at preventing symptoms, maintaining individual mobility, and strengthening bone and muscle. Overall physical and psychological well-being and Nutrition is also very important
Diet:
Diet should include adequate intake of calcium and vitamin D adjusted for the diminished weight of most patients
Bisphosphonate & other medicines:
Bisphosphonate therapy (I.V. pamidronate or zolendronate) is commonly used to treat children with OI who have frequent fractures, spinal compression fractures, bone pain and decreased bone density measured by DEXA scan.
Bisphosphonates work by slowing down the resorption of existing bone while new bone is being formed. This allows bone mass and strength to increase. However, it does not make the new bone normal.
Adults with OI may be treated with oral or intravenous bisphosphonates.
Other drugs:
Denosumab decreases bone resorption
Teriparatide has been shown to increase bone strength
Surgical management:
Surgery is needed in patients where there is progressive deformity of a bone or if a bone fractures repeatedly
Metal rods are placed into the long bones of the upper and lower extremities
Surgery to relieve compression between the base of the skull and the top of the spine (basilar invagination) may prove necessary in severe symptomatic patients
Dental management:
Treatments, including capping teeth, braces, and surgery may be needed
Successful implant placement has been reported in a few cases, although the impact of the altered bone on osseointegration is not well studied.
In patients with significant malocclusion, orthognathic surgery and orthodontic treatment may be performed.
Alternatively, osteodistraction may be considered to reduce the risk of atypical fractures from conventional orthognathic procedures (e.g., Le Fort I osteotomy)
Physical and occupational therapy:
Both are very important in babies and children with OI.
Assistive devices:
Wheelchairs and other custom-made equipment may be needed as babies get older.
Reference
Harrison’s Principles of Internal Medicine, 20th Edition
NEVILLE’S – Oral & Maxillofacial Pathology 4E
Robbins & Cotran – Pathologic Basis of Disease
National Organization of Rare Disorders
A Visual Learning Platform



